中小学合成材料面层运动场地新国标GB 36246-2018
Nov 24, 2018 /乐邦塑胶跑道
由于本文章比较长,如果需要简单了解塑胶跑道新国标新的内容,可以访问:新国标塑胶跑道到底“新”到哪里?
需要下载全文的,百度网盘:链接: https://pan.baidu.com/s/1E2YZ_cABfwREyhmE_3QUCQ 密码: gsa2

前 言
本标准的全部技术内容为强制性。
本标准按照 GB/T 1.1-2009 给出的规则起草。
本标准代替 GB/T 19851.11-2005《中小学体育器材和场地 第 11 部分:合成材料面层运动场地》。与 GB/T 19851.11-2005 相比,除编辑性修改外主要技术变化如下:
——增加了合成材料面层、现浇型面层、预制型面层、人造草面层、渗水型面层、非渗水型面层、固体原料、非固体原料、厚度、冲击吸收、垂直变形、拉伸强度、拉断伸长率、抗滑值、耐老
化性能、阻燃性能、总挥发性有机化合物、挥发性有机化合物的术语和定义,删除了合成面层、平整度、压缩复原率、1 级阻燃的术语和定义(见第 3 章, 2005 年版的第 3 章);
——增加了分类(见第 4 章);
——增加了铺装要求(见 5.1);
——修改了厚度要求(见 5.2,2005 年版的 4.4);
——删除了外观和规格、标志线、平整度的要求(见 2005 年版的 4.1、4.2、4.3);
——增加了物理机械性能中冲击吸收、垂直变形、抗滑值项目及指标要求,删除了硬度(邵 A)、压
缩复原率、回弹值性能要求,修改了拉伸强度项目及指标要求,修改了拉断伸长率及其指标要
求(见 5.3.1,2005 年版的 4.6);
——修改了厚度、拉伸强度、拉断伸长率、阻燃性能的试验方法(见 6.1、6.4、6.6,2005 年版的
附录 B、5.6 和附录 E);
——增加了冲击吸收、垂直变形和抗滑值的试验方法(见附录 D、附录 E 和附录 F);
——增加了人造草面层的物理机械性能要求和试验方法(见 5.3.2、6.2、6.3、6.7、6.8);
——增加了合成材料面层耐老化性能要求及试验方法(见 5.4、6.9);
——增加了除人造草面层以外的合成材料面层中无机填料含量要求及试验方法(见 5.5.1、6.10);
——增加了合成材料面层防滑胶粒及人造草面层填充合成材料颗粒中高聚物总量要求及试验方法
(见 5.5.2、6.11);
——增加了合成材料面层运动场地成品和原料中有害物质限量要求和气味要求及试验方法(见
5.6、附录 A、附录 B、附录 G、附录 H、附录 I、附录 J 和 6.12、6.13、6.14、6.15);
——增加了取样要求(见第 7 章);
——增加了检验规则(见第 8 章)。
本标准由中华人民共和国教育部提出并归口。
本标准起草单位:教育部教育装备研究与发展中心、清华大学、中国建材检验认证集团股份有限公
司、深圳市计量质量检测研究院、上海建科检验有限公司、中国环境科学研究院、南京林业大学、山东
省产品质量检验研究院、浙江省家具与五金研究所、环境保护部固体废物与化学品管理技术中心、广州
同欣康体设备有限公司、江苏省产品质量监督检验研究院、广州质量监督检测研究院、上海市化学建材
行业协会、武汉体育学院、中国聚氨酯工业协会、国正检验认证有限公司、国家体育总局体育器材装备
中心、山东泰山体育工程有限公司、上海航宽体育场设施工程有限公司、广州大洋元亨化工有限公司、
都佰城新材料技术(上海)有限公司、广州杰锐体育设施有限公司、深圳市领先康体实业有限公司、麦
迪人造草坪股份有限公司、青岛科兴教育装备有限公司。
本标准主要起草人:张彭义、李平、郭中宝、吴海涛、车燕萍、党建伟、余若祯、罗振扬、蔡陈敏、
丁琼、黄金飞、朱晓翠、张耀东、张滨、王微山、孟庆生、楼明刚、李建波、郑伟涛、陈韶、侯力波、
吴颖、陈琳、黄宇梁、王港、贾志勇、王季安、余询、周立成、郭飞、卞志勇、李志实、郭龙、师建华、
林凡秋、吴雪、孙震。
本标准所代替标准的历次版本发布情况为:
——GB/T 19851.11-2005。
中小学合成材料面层运动场地
1 范围
本标准规定了中小学合成材料面层运动场地的术语和定义、分类、技术要求、试验方法、取样要求
及检验规则。
本标准适用于中小学校新建、改建和扩建的室外合成材料面层运动场地的设计、选材、铺装、检测
与验收。
2 规范性引用文件
下列文件对于本文件的应用是必不可少的。凡是注日期的引用文件,仅注日期的版本适用于本文件。
凡是不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T 4498.1-2013 橡胶 灰分的测定 第 1 部分:马弗炉法
GB/T 6682-2008 分析实验室用水规格和试验方法
GB/T 10111-2008 随机数的产生及其在产品质量抽样检验中的应用程序
GB/T 10654-2001 高聚物多孔弹性材料 拉伸强度和拉断伸长率的测定
GB/T 14833-2011 合成材料跑道面层
GB/T 14837.1 橡胶和橡胶制品 热重分析法测定硫化胶和未硫化胶的成分 第 1 部分:丁二烯橡
胶、乙烯-丙烯二元和三元共聚物、异丁烯-异戊二烯橡胶、异戊二烯橡胶、苯乙烯-丁二烯橡胶
GB/T 14837.2 橡胶和橡胶制品 热重分析法测定硫化胶和未硫化胶的成分 第 2 部分:丙烯腈-
丁二烯橡胶和卤化丁基橡胶
GB/T 16422.2-2014 塑料 实验室光源暴露试验方法 第 2 部分:氙弧灯
GB/T 16483 化学品安全技术说明书 内容和项目顺序
GB/T 18204.2 公共场所卫生检验方法 第 2 部分:化学污染物
GB/T 18446 色漆和清漆用漆基 异氰酸酯树脂中二异氰酸酯单体的测定
GB 18581-2009 室内装饰装修材料 溶剂型木器涂料中有害物质限量
GB 18583 室内装饰装修材料 胶粘剂中有害物质限量
GB/T 18883-2002 室内空气质量标准
GB/T 22517.6 体育场地使用要求及检验方法 第 6 部分:田径场地
GB/T 23986-2009 色漆和清漆 挥发性有机化合物(VOC)含量的测定 气相色谱法
GB/T 23991 涂料中可溶性有害元素含量的测定
HJ 865-2017 恶臭嗅觉实验室建设技术规范
QB/T 1090-2001 地毯绒簇拔出力的试验方法
ISO 16000-3 室内空气 第 3 部分:室内空气和试验室空气中甲醛与其他羰基化合物的测定主动
取样法(Indoor air - Part 3: Determination of formaldehyde and other carbonyl compounds in
indoor air and test chamber air - Active sampling method)
ISO 16000-6 室内空气 第 6 部分:通过 Tenax TA 吸附剂、热解吸以及使用质谱(MS)或质谱-火
焰离子化检测器(MS-FID)的气相色谱主动取样来测定室内和试验室空气中的挥发性有机化合物(Indoor
air - Part 6: Determination of volatile organic compounds in indoor and test chamber air by
active sampling on Tenax TA sorbent, thermal desorption and gas chromatography using MS or
MS-FID)
3 术语和定义
GB/T 10654-2001、GB/T 14833-2011、GB/T 18581-2009 和 GB/T 18883-2002 界定的以及下列术语
和定义适用于本文件。为了便于使用,以下重复列出了 GB/T 10654-2001、GB/T 14833-2011、GB/T
18581-2009 和 GB/T 18883-2002 中的某些术语和定义。
3.1
合成材料面层 synthetic surface
铺装在沥青混凝土或水泥混凝土等基础层上的高分子合成材料层。
3.2
现浇型面层 in-situ casting surface
将高分子原料和其他原料在现场浇注铺装的面层。
3.3
预制型面层 prefabricated surface
按一定的生产工艺流程将高分子合成材料预先制备成一定厚度的卷材或块材,至现场粘结或拼装的
面层。
3.4
人造草面层 artificial turf surface
以类似天然草的合成纤维经机械编织固定于底布层上所形成的合成材料面层。
3.5
渗水型面层 permeable surface
由树脂粘合橡胶碎粒或其他方法制造的具有缝隙结构的一类合成材料面层。水在该类型面层上除存
在表面径流外,还存在通过面层的流动形式。
注:改写自 GB/T 14833-2011 中 3.2.1。
3.6
非渗水型面层 non-permeable surface
垂直剖面致密或有少量气孔及带有特定结构形式的一类合成材料面层。
注:改写自 GB/T 14833-2011 中 3.2.2。
3.7
固体原料 solid raw materials
在铺装时以固体形式存在的合成材料。
注:如丁苯橡胶颗粒、三元乙丙橡胶颗粒、聚氨酯橡胶颗粒、热塑性弹性体、预制卷材、人造草等。
3.8
非固体原料 non-solid raw materials
在铺装时以非固体形式存在的合成材料。
注:如各种胶粘剂、现浇型面层用预聚体和多元醇树脂组分等。
3.9
厚度 thickness
合成材料面层表面与其底面之间的总垂直距离。
3.10
冲击吸收 shock absorbency
合成材料面层对冲击力的减缓性能。
3.11
垂直变形 vertical deformation
20 kg 重物以规定的高度落在合成材料面层时,合成材料面层在垂直方向的变形。
3.12
拉伸强度 tensile strength
拉伸试样到断裂所施加的最大拉伸应力。
[GB/T 10654-2001,定义3.1]
3.13
拉断伸长率 elongation at break
断裂试样的百分伸长率。
[GB/T 10654-2001,定义3.2]
3.14
抗滑值 anti-skidding value
合成材料面层在干燥或潮湿状态下通过滑动摩擦阻力吸收能量的程度。
3.15
耐老化性能 aging resistance
合成材料面层具有的推迟延缓老化的性质。
3.16
阻燃性能 flame retardance
合成材料面层具有的推迟火焰蔓延的性质。
3.17
总挥发性有机化合物 total volatile organic compounds (TVOC)
利用 Tenax GC 或 Tenax TA 采样,非极性色谱柱(极性指数小于 10)进行分析,保留时间在正己
烷和正十六烷之间的挥发性有机化合物。
[GB/T 18883-2002,定义 3.3]
3.18
挥发性有机化合物 volatile organic compounds (VOC)
在 101.3 kPa 标准大气压力下,任何初沸点低于或等于 250 ℃的有机化合物。
[GB 18581-2009,定义 3.1]
4 分类
4.1 合成材料面层运动场地按使用功能分为田径场地、球类场地和其他活动场地。
4.2 合成材料面层按材料形态分为现浇型面层、预制型面层和人造草面层。
5 技术要求
5.1 铺装要求
5.1.1 合成材料面层的铺装应综合评估场地及其周边的通风、扩散条件,应有利于挥发性有机化合物
的散发,并避免铺装时废气、废水、固体废弃物等对场地及周边环境的污染。
5.1.2 铺装前应提供所需使用的原料清单(包括品名和数量)、按照 GB/T 16483 编写的化学品安全技
术说明书和型式检验报告,所使用的原料以及铺装后的运动场地在正常及预期使用条件下不应对人体健
康和生态环境产生危害。
5.1.3 不应使用煤焦油沥青作为场地基础材料。
5.2 厚度
现浇型和预制型面层厚度应符合表 1 规定。
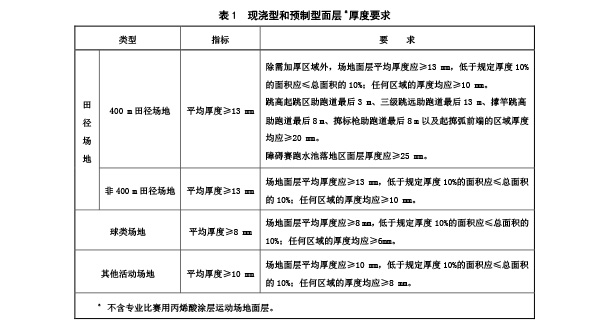
5.3 物理机械性能
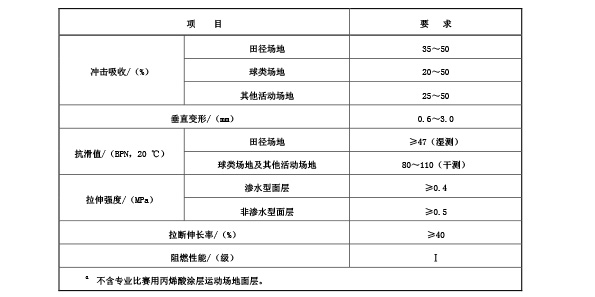
5.3.1 现浇型和预制型面层物理机械性能应符合表2规定。
表 2 现浇型和预制型面层
a 物理机械性能要求
5.3.2 人造草面层成品及草丝的物理机械性能应符合表 3 规定。
表 3 人造草面层成品及草丝的物理机械性能要求

5.4 合成材料面层耐人工气候老化性能
5.4.1 现浇型和预制型面层加速老化 500 h 后,拉伸强度和拉断伸长率应符合表 2 要求。
5.4.2 人造草面层草丝加速老化 500 h 后,草丝拉断力应不低于加速老化前测定值的 80%。
5.5 合成材料面层中无机填料及高聚物的含量
5.5.1 除人造草面层以外的合成材料面层中,无机填料含量应 ≤ 65%。
5.5.2 合成材料面层防滑胶粒及人造草面层填充用合成材料颗粒中高聚物总量应 ≥ 20%。
5.6 合成材料面层成品和原料中有害物质限量及气味
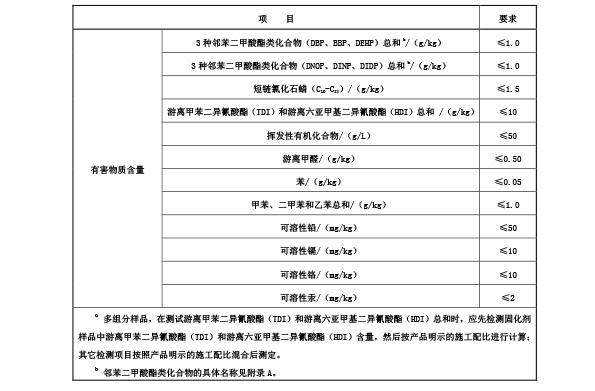
5.6.1 合成材料面层成品中有害物质限量及气味
现浇型和预制型面层成品中有害物质限量及气味应符合表? 4? 要求,人造草面层成品中有害物质限量
应符合表? 5? 要求。
表? 4 现浇型和预制型面层成品中有害物质限量及气味要求

表 5 人造草面层成品中有害物质限量要求
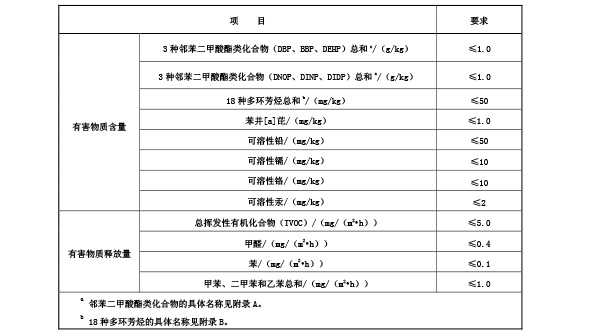
表 5 人造草面层成品中有害物质限量要求
5.6.2 合成材料面层原料中有害物质限量及气味
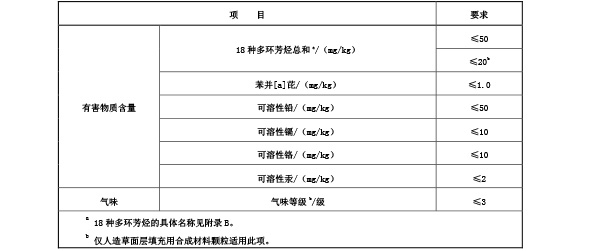
5.6.2.1 铺装时使用的固体原料(包括防滑颗粒、填充颗粒、铺装前的预制型面层和人造草等)中有
害物质限量及气味要求应符合表 6 要求。
表 6 固体原料中有害物质限量及气味要求
5.6.2.2 铺装时使用的非固体原料(包括各种胶粘剂、现浇型面层用预聚体和多元醇树脂组分等)中
有害物质限量应符合表 7 的要求。
表 7 非固体原料中有害物质限量要求a
6 试验方法
6.1 厚度的测定
6.1.1 田径场地按 GB/T 22517.6 规定的方法进行。
6.1.2 球类场地及其他活动场地按附录 C 的规定进行。
6.2 冲击吸收的测定
按附录 D 的规定进行。
6.3 垂直变形的测定
按附录 E 的规定进行。
6.4 拉伸强度、拉断伸长率的测定
按 GB/T 10654-2001 规定的方法进行。
6.5 抗滑值的测定
按附录 F 的规定进行。
6.6 阻燃性能的测定
按 GB/T 14833-2011 规定的方法进行。
6.7 草丝拉断力的测定
6.7.1 试样制备
在不同行任意选择5簇草丝,从每簇草丝中任选一根草丝进行拉伸试验。
6.7.2 试验程序
在精度为1 N以上的拉力试验机上进行试验,试验速度为(250±50) mm/min,记录试样断裂时的拉
断力示值,5个试样试验,取结果的算术平均值,精确到1 N。
注:如果某个试样的试验结果与该批试样平均值的允差超过30%,则该试样试验无效,重新选择一根草丝进行试验。
6.8 单簇草丝拔出力的测定
按照QB/T 1090-2001的规定,利用器具夹住一个单簇草丝的端头,从人造草的结构中向上拔出,并
在拔出过程中记录所示的最大力值。
6.9 耐老化性能的测定
按 GB/T 16422.2-2014 的规定进行氙灯辐照试验,试验条件为方法 A、循环序号 1,试验 500 h 后,
按 6.4 测定拉伸强度、拉断伸长率,按 6.7 测定草丝拉断力。
6.10 无机填料含量的测定
按照 GB/T 4498.1-2013 方法 A 的规定进行,试验温度(550±25)℃,测得的灰分含量作为无机填
料含量。
6.11 高聚物总量的测定
按照 GB/T 14837.1 和 GB/T 14837.2 的规定进行。
6.12 合成材料面层成品和固体原料中有害物质含量的测定
6.12.1 样品前处理
取样品适量,采用冷冻研磨或不致产生热量的其他加工方式粉碎,选取粒径在 0.85 mm~1.40 mm
(20 目~14 目)之间的细小颗粒,作为检测用试样。制样过程应避免使用不锈钢研磨装置以防止其造
成的铬等污染。如样品颗粒粒径小于 1.40 mm,可直接作为检测用试样。检测各项二异氰酸酯含量的试
样在样品前处理过程中应避免空气中水分的影响。
对于现场挖取的样品,制样前应去除底层附着物,以避免基础层对面层材料可能的污染。
除重金属外,其余项目应在样品前处理后立即检测。
6.12.2 测定
6.12.2.1 邻苯二甲酸酯类化合物(DBP、BBP、DEHP、DNOP、DINP、DIDP)的测定按附录 A 中的规定进
行。
6.12.2.2 18 种多环芳烃总和及苯并[a]芘的测定按附录 B 中的规定进行。
6.12.2.3 短链氯化石蜡(C 10 -C 13 )的测定按附录 G 中的规定进行。
6.12.2.4 4,4'-二氨基-3,3'-二氯二苯甲烷(MOCA)的测定按附录 H 中的规定进行。
6.12.2.5 游离甲苯二异氰酸酯(TDI)和游离六亚甲基二异氰酸酯(HDI)总和的测定按 GB/T 18446
中的规定进行。
6.12.2.6 游离二苯基甲烷二异氰酸酯(MDI)的测定按 GB/T 18446 中的规定进行。
6.12.2.7 可溶性铅、镉、铬、汞的测定按 GB/T 23991 中的规定进行。
6.13 合成材料面层成品中有害物质释放量的测定
按附录 I 中的规定进行。
6.14 气味评定
按附录 J 中的规定进行。
6.15 合成材料面层非固体原料中有害物质含量的测定
6.15.1 游离甲醛的测定按 GB 18583 中的规定进行。
6.15.2 苯、甲苯/二甲苯/乙苯总和的测定按 GB 18581 中的规定进行。
6.15.3 游离甲苯二异氰酸酯(TDI)和游离六亚甲基二异氰酸酯(HDI)总和的测定按 GB/T 18446 中
的规定进行。
6.15.4 邻苯二甲酸酯类化合物的测定,称取(0.2~0.3)g(精确至 0.1 mg)试样置于 50 mL 的容量
瓶中,用乙酸乙酯溶解/分散并定容,摇匀,然后按照附录 A 中的规定进行分析。
6.15.5 短链氯化石蜡(C 10 -C 13 )的测定按照附录 G 中的规定进行。
6.15.6 挥发性有机化合物含量的测定按 GB/T 23986-2009 的规定进行。其中,样品称量为 1 g(精确
至 0.1 mg),按方法 2 计算挥发性有机化合物含量,多组分原料按明示配比进行混合,搅拌均匀后称
量。
6.15.7 可溶性铅、镉、铬、汞的测定按 GB/T 23991 中的规定进行。
7 取样要求
7.1 见证取样
验收检验样品应在建设方(或代建方、使用方)、监理方及施工方代表等相关人员见证下在铺装现
场取样。
7.2 合成材料面层原料样品
7.2.1 应对每次进场的原料取样,同一批次同一规格原料取一组样品。非固体原料每组取样量不少于
250 mL,多组分非固体原料按配比取样,配比最小的组分取样量应不少于 50 mL。预制型面层和人造草
面层样品规格不小于 300 mm×400 mm×实际厚度,其他固体原料每组取样量不少于 500 g。
7.2.2 非固体原料在充分搅拌均匀后装入洁净干燥的玻璃瓶或其他不会导致化学污染的容器中密封保
存,多组分非固体原料应将各组分单独取样包装。固体原料取样后装入聚乙烯或聚四氟乙烯袋密封保存。
7.3 合成材料面层成品样品
7.3.1 样品规格及取样位置
铺装现场裁取、挖取或平行制备的合成材料面层样品规格不小于 300 mm×400 mm×实际厚度,取
样后装入聚乙烯或聚四氟乙烯袋密封保存。运动场地上挖取样品的位置应按附录 K 确定。
7.3.2 现浇型面层样品
物理机械性能及无机填料含量检测用样品应在合成材料面层现场铺装的同时平行制备,平行样的制
备配方、工艺和厚度应与现场施工相同;样品数量不少于 3 块,其中 1 块作为检测用样,其余作为复验
备样。必要时,可在铺装完成后的场地上挖取样品。
有害物质限量及气味检测用样品应在合成材料面层铺装后 14 d~28 d 内直接从运动场地上挖取 1
块样品。
7.3.3 预制型面层和人造草面层样品
物理机械性能及无机填料和高聚物含量检测用样品应在现场裁取未铺装的合成材料面层,取样数量
不少于 3 块,其中 1 块作为检测用样,其余作为复验备样。人造草面层填充颗粒取样量按取样面积与单
位面积颗粒填充量计算确定。必要时,应在铺装完成后的运动场地上挖取样品,挖取的人造草面层样品
应不带胶粘剂。
有害物质限量及气味检测用样品应在合成材料面层铺装完成后 14 d~28 d 内直接从运动场地上挖
取 1 块样品。铺装后现场挖取的预制型面层样品按合成材料面层成品的要求进行检验;铺装后现场挖取
的人造草面层样品中的填充颗粒按合成材料面层固体原料的要求进行检验,去除填充颗粒后的人造草面
层按合成材料面层成品的要求进行检验。
7.4 运输、保存与检测时间
样品运输过程中应避免因扭曲、挤压、受潮、化学污染或高温等改变样品物理或化学完整性,样品
送达实验室后应在温度为(25±5)℃的室内环境带包装保存,原料样品应在送达实验室后 14 d 内开始
检测,成品样品应在合成材料面层铺装完毕后 14 d~60 d 内开始检测。
8 检验规则
8.1 验收检验
验收检验项目为 5.2、5.3、5.5、5.6。
8.2 型式检验
型式检验项目为 5.2~5.6,出现下列情况之一时,合成材料面层产品应进行型式检验:
a) 新产品、新工艺、新配方定型鉴定时;
b) 正常生产后,原材料、生产工艺、配方有重大变化时;
c) 停产或未施工六个月及以上恢复生产或施工时;
d) 正常生产或施工时,每年进行 1 次;
e) 质量主管部门监督抽查时。
8.3 检验结果的判定
经检验,检验项目全部合格,判定所检样品符合本标准要求;检验项目中任一项或一项以上不合格,
判定所检样品不符合本标准要求。
8.4 复验规则
8.4.1 原料样品经检验不符合本标准要求的,可另取双倍样品进行复验,检验项目全部合格,判定所
检样品复验符合本标准要求,否则判定不符合本标准要求。
8.4.2 成品样品经初次检验如有部分指标不符合本标准要求的,可以安排复验。如物理机械性能、无
机填料含量或高聚物总量存在不合格项目,可对复验备样进行检测,如果 2 块复验备样全部合格,判定
复验符合本标准要求,否则判定不符合本标准要求;必要时,可在铺装完成后的场地上现场见证挖取样
品进行复验,如检验项目全部合格,判定复验符合本标准要求,否则判定不符合本标准要求。如有害物
质或气味要求存在不合格项目,可在 60 d 内整改完毕后重新在铺装完成后的场地上现场见证挖取样品
进行检验,如检验项目全部合格,判定复验符合本标准要求,否则判定不符合本标准要求。
8.5 场地符合性判定原则
所有检验项目,包括复验项目(如有)均符合本标准要求时,判定该场地符合本标准要求。
附 录 A
(规范性附录)
邻苯二甲酸酯类化合物含量的测试 气相色谱-质谱法
A.1 概述
本方法规定了用气相色谱-质谱法测定合成材料面层及其原料中邻苯二甲酸酯类化合物含量的方
法。
A.2 原理
试样用乙酸乙酯或者其他合适的溶剂进行超声萃取,试样溶液冷却后用气相色谱-质谱联用仪测定,
采用总离子流色谱图(TIC)进行定性,选择离子检测(SIM)进行定量。
A.3 材料和试剂
A.3.1 萃取溶剂
乙酸乙酯或者其他合适溶剂,分析纯。
A.3.2 校准化合物
邻苯二甲酸二丁酯(DBP)、邻苯二甲酸丁苄酯(BBP)、邻苯二甲酸二(2-乙基己基)酯(DEHP)、
邻苯二甲酸二正辛酯(DNOP)、邻苯二甲酸二异壬酯(DINP)、邻苯二甲酸二异癸酯(DIDP),纯度≥
98%或已知纯度。
A.3.3 标准储备溶液
分别称取适量的邻苯二甲酸酯类标准品,用萃取溶剂(A.3.1)配制成 DBP、BBP、DEHP、DNOP 浓度
为 200 mg/L,DINP、DIDP 浓度为 500 mg/L 的混合标准储备溶液。标准储备溶液宜在(0~4)℃冰箱中
保存,配制后 180 d 内使用。
A.3.4 标准工作溶液
将标准储备溶液采用逐级稀释的方法配制 DBP、BBP、DEHP、DNOP 浓度从 0.5 mg/L 到 10 mg/L,DINP、
DIDP 浓度从 2.5 mg/L 到 50 mg/L 之间的不少于 5 点的混合标准工作溶液。标准工作溶液宜在(0~4)℃
冰箱中保存,配制后 90 d 内使用。
A.3.5 有机相微孔滤膜
孔径 0.45 μ m。
A.4 仪器设备
A.4.1 气相色谱-质谱联用仪。
A.4.2 超声波发生器。
A.4.3 电子天平:精确度0.1 mg。
A.5 分析步骤
A.5.1 气相色谱-质谱联用仪工作条件
由于测试结果取决于所使用的仪器,因此不可能给出仪器分析的普适参数,可参考的仪器条件如
下:
a) 色谱柱:5%苯基-甲基硅氧烷毛细管柱,30 m×0.25 mm×0.25 μ m;
b) 进样口温度:290 ℃;
c) 柱温:程序升温,50 ℃保持2 min,然后以20 ℃/min 升至200 ℃再以8 ℃/min 升至300 ℃
保持5.5 min;
d) 质谱接口温度:280 ℃;
e) 离子源温度:230 ℃;
f) 电离方式:EI;
g) 电离能量:70 eV;
h) 质量扫描范围:50 amu~500 amu;
i) 测试方式:全扫描的总离子流图(TIC)定性,选择离子监测(SIM)定量;
j) 进样方式:不分流进样;
k) 载气:氦气(纯度≥99.999%),流量为1.0 mL/min;
l) 进样量:1.0 μ L;
m) 溶剂延迟:5 min。
A.5.2 试样制备
称取约0.5 g样品,精确至0.1 mg,放入螺口刻度试管(带密封盖),加入10 mL萃取溶剂(A.3.1)
并密封试管,置于超声波发生器(A.4.2)中,在60 ℃水温下超声萃取60 min。萃取完成后,取出刻度
试管冷却至室温并混合均匀,用微量注射器取l μ L进样,测其峰面积。若试样溶液的峰面积大于A.3.4
中最大浓度的峰面积,则用萃取溶剂(A.3.1)稀释后测定,并将稀释倍数f代入计算公式中。
注1:高浓度样品可进行两次萃取测试。
注2:如果待测溶液中有颗粒物质,取一部分试液经0.45μ m有机系微孔滤膜过滤后用于检测分析。
A.5.3 定性分析
进行样品测试时,如果检出色谱峰的保留时间与标准样品相一致,并且在扣除背景后样品质谱图中
所有选择离子均出现,而且其丰度比与标准品的丰度比相一致(相对丰度>50%,允许±10%的偏差;相
对丰度在20%~50%之间,允许±15%的偏差;相对丰度在10%~20%之间,允许±20%的偏差;相对丰度
≤10%,允许±50%的偏差),则可判断样品中存在相应的邻苯二甲酸酯类。
在A.5.1条件下,6种邻苯二甲酸酯类化合物的特征离子及其丰度比见表A.1。
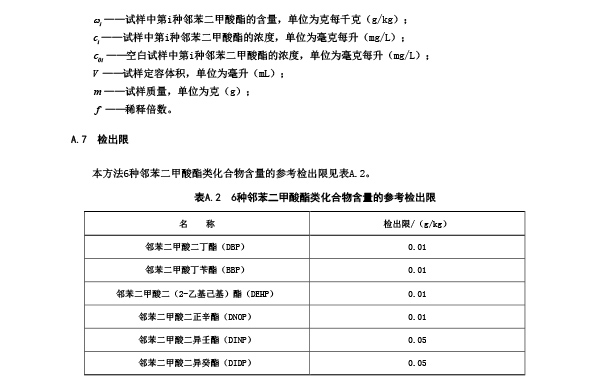
表 A.1 6 种邻苯二甲酸酯类化合物的定性参考离子和定量选择离子
A.5.4 定量分析
A.5.4.1 标准溶液和试液中待测试的邻苯二甲酸酯类化合物的响应值均应在仪器检测的线性范围内,
如果试液的检测响应值超出仪器检测的线性范围,可适当稀释后测试。
A.5.4.2 本标准采用外标法对邻苯二甲酸酯类化合物进行定量分析。在色谱图中,选取适当的定量选
择离子(参见表 A.1)进行峰面积积分,DINP 和 DIDP 应分别将其所有同分异构体的色谱峰的基线拉平
后积分,计算其面积的总和,按式(A.1)计算样品中每种邻苯二甲酸酯的含量。
A.5.4.3 DINP 和 DIDP 由于包含不可分离的同分异构体,出峰存在部分重叠,并且如果同时存在 DNOP,
在色谱图上 DNOP 出峰也会与 DINP 出峰出现重叠。因此在选取定量离子时应避免 DNOP、DINP 和 DIDP
之间的相互干扰,DNOP 选择 m/z=279、DINP 选择 m/z=293、DIDP 选择 m/z=307可在最大程度上减少相互
之间的干扰。
A.5.5 空白试验
除不加试样外,均按照上述分析步骤进行。
A.6 结果计算
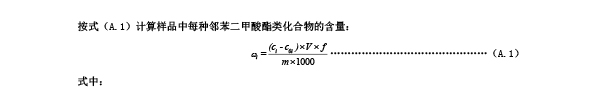
A.8 精密度
同一实验室相同条件下,在短时间内对同一被测对象,两次独立测定结果的绝对差值不应大于
其算术平均值的10%。
附 录 B
(规范性附录)
18 种多环芳烃含量的测试 气相色谱-质谱法
B.1 概述
本方法规定了用气相色谱-质谱法测定合成材料面层及其原料中 18 种多环芳烃含量的方法。
B.2 原理
试样用乙酸乙酯或者其他合适的溶剂进行超声萃取, 萃取液经冷却后试样经超声波水浴提取,
提取液冷却后为待测溶液,用气相色谱-质谱联用仪测试,内标法定量。
B.3 材料和试剂
B.3.1 萃取溶剂
乙酸乙酯或其他合适的溶剂,均为分析纯。
B.3.2 标准工作溶液
18种多环芳香烃(PAHs)混合标准溶液。
B.3.3 内标溶液
内标溶液如下:
a) 内标物 1:八氘代萘溶液(naphthalene-d 8 ),用于校正表 B.1 中 2 号 PAH;
b) 内标物 2:十氘代芘溶液(pyrene-d 10 ),或十氘代蒽溶液(anthracene-d 10 ),或十氘代菲溶
液(Phenanthrene-d 10 ),用于校正表 B.1 中 3 号~8 号,10 号~12 号 PAHs;
c) 内标物 3:十二氘代苝溶液(perylene-d 12 ),或十二氘代苯并[a]芘溶液[benzo(a)pyrene-d 12 ],
三苯基苯(triphenylbenzene),用于校正表 B.1 中 13 号~17 号,19 号~21 号 PAHs。
B.3.4 内标物混合溶液
移取适量内标物 1、内标物 2、内标物 3,用萃取溶剂(B.3.1)稀释成含内标物 l 质量浓度为 0.3 mg/L、
内标物 2 质量浓度为 0.42 mg/L、内标物 3 质量浓度为 0.96 mg/L 的混合内标液,至少应使用 3 种内标
物质。
B.3.5 混合标准溶液配制
移取适量 18 种 PAHs 混合标准溶液(B.3.2),用萃取溶剂(B.3.1)逐级稀释成质量浓度为 0.1 mg/L、
0.05 mg/L、0.02 mg/L、0.01 mg/L、0.002 mg/L 的系列标准溶液各 1 mL,再分别加入 100 μ L 内标物
混合溶液(B.3.4),混合均匀。
B.3.6 有机相微孔滤膜
孔径 0.45 μ m。
B.4 仪器设备
B.4.1 气相色谱-质谱联用仪。
B.4.2 超声波发生器。
B.4.3 电子天平:精确度0.1 mg。
B.5 分析步骤
B.5.1 样品制备
称取 0.5 g 样品,精确至 0.1 mg,放入螺口刻度试管(带密封盖),加入 10 mL 萃取溶剂(B.3.1)
并密封试管,置于超声波水浴装置中,在 60 ℃水温下超声萃取 60 min。萃取完成后,取出刻度试管冷
却至室温并混合均匀,得到的试样溶液可依据其实际情况直接进样,或者用萃取溶剂(B.3.1)稀释后
用于测试。
移取 1 mL 待测液,加入 100 μ L 内标物混合溶液(B.3.4)混匀后进行气相色谱-质谱分析。
注 1:高浓度样品可进行两次萃取测试。
注 2:如果试样溶液中有颗粒物质,取一部分试液经 0.45μ m 有机系微孔滤膜过滤后用于检测分析。
B.5.2 内标标准曲线的绘制
对标准混合溶液(B.3.5)进行气相色谱-质谱分析,以待测物的质量浓度为横坐标、待测物和对应
内标物峰面积的比值为纵坐标作图,可得一条通过原点的直线,即内标标准曲线。
B.5.3 气相色谱-质谱分析条件
由于测试结果取决于所使用的仪器,因此不可能给出仪器分析的普适参数,可参考的仪器条件如下:
a) 色谱柱:5%苯基-甲基硅氧烷毛细管柱,30 m×0.25 mm×0.25 μ m;
b) 进样口温度:280 ℃;
c) 柱温:程序升温,50 ℃保持 2 min,然后以 20 ℃/min 升至 200 ℃再以 8 ℃/min 升至 300 ℃
保持 5.5 min;
d) 质谱接口温度:280 ℃;
e) 离子源温度:270 ℃;
f) 电离方式:EI;
g) 电离能量:70 eV;
h) 质量扫描范围:45 aum~350 aum;
i) 测试方式:选择离子监测(SIM);
j) 进样方式:不分流进样;
k) 载气:氦气(纯度≥99.999%),流量为 1.0 mL/min;
l) 进样量:1.0 μ L;
m) 溶剂延迟:5 min。
B.5.4 定性及定量分析
根据各实验室仪器所适合的分析条件对混合标准溶液及待测液进行分析,根据色谱峰的保留时间
和特征离子的相对丰度进行定性分析。以下条件可用于判定样品中是否含有多环芳烃:
a) 样品中目标物保留时间与标液中目标物保留时间的偏差在±0.5%或±0.1 min范围内;
b) 特征离子在标液中目标物的保留时间处出峰;
c) 特征离子的相对丰度与标液中目标物的相对丰度一致(相对丰度>50%,允许±10%的偏差;
相对丰度 20%~50%之间,允许±15%的偏差;相对丰度 10%~20%之间,允许±20%的偏差;
相对丰度≤10%,允许±50%的偏差)。
定量分析参考表 B.1 中的定量离子,采用内标法定量。
表 B.1 18 种多环芳烃与内标物定性参考离子和定量选择离子
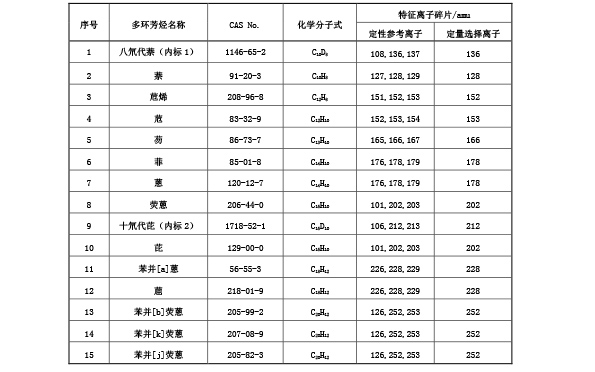
表? B.1 18? 种多环芳烃与内标物定性参考离子和定量选择离子(续)

将按式(B.2)计算得到的每种多环芳烃的含量加和即得到试样中18种多环芳烃总和。
表? B.1 18? 种多环芳烃与内标物定性参考离子和定量选择离子(续)
将按式(B.2)计算得到的每种多环芳烃的含量加和即得到试样中18种多环芳烃总和。
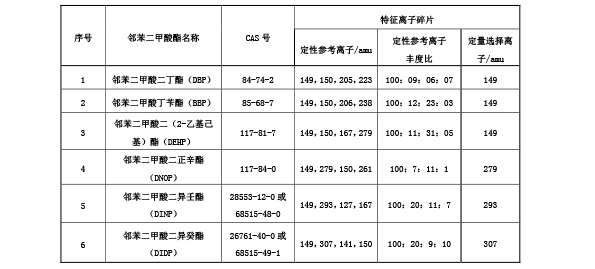
B.7 检出限
本方法单个多环芳香烃化合物的参考检出限为 0.1 mg/kg。
B.8 精密度
同一实验室相同条件下,在短时间内对同一被测对象,两次独立测定结果的绝对差值不应大于其算
术平均值的15%。
附 录 C
(规范性附录)
球类场地及其他活动场地面层厚度的测量方法
C.1 测量仪器
三针测厚仪,精度为 1 mm。
C.2 测量方法
每个场地至少取 12 个点,在场地端线开始向对侧测量 3 排点,每排 4 个测试点,视场地长短均匀
排定。把三针测厚仪放在场地上,手持专业测厚仪垂直向下,压向运动地面直至基础层表面即停止,专
业测厚仪上相对应的数字即为测量厚度。
C.3 结果计算
C.3.1 取各个测试点的测量厚度的平均值为平均厚度,数值精确到小数点后一位。
C.3.2 低于规定厚度10%的面积占总面积的百分比按式(C.1)进行计算:

附 录 D
(规范性附录)
合成材料面层冲击吸收的检测方法
D.1 检测装置
D.1.1 采用冲击吸收测试仪(见图 D.1 所示)检测合成材料面层的冲击吸收性能。

e) 金属导向管,其内沿内径是(71.0±0.1) mm;
f) 测力脚,是由钢制测力台,压力传感器,弹簧和铁砧(最低厚度 20 mm,以盘中心的测量值为
准)组成的,固定在金属导向管中,整体质量(不计导向管)应该为(3.0±0.3)kg;
g) 提升与释放重物装置,可以让其从设定的高度跌落,且误差不大于±0.25 mm;
h) 具有记录和调节放大从压力传感器输出的信号的装置以及显示这些记录的装置;
i) 电子测量记录装置装有放大器和记录放大器,以及过滤性很低的过滤器,并能在 0.01 s 内记
录单个冲击产生的力的最大值,精确度为 0.5%,放大器的频道频率应该大于等于 1kHz;
j) 测试地基应该是一块平整、坚硬、无振动的混凝土地板,可得到 D.3 中要求的
C
F 数据。
D.2 检测方法
将质量为 20 kg的下落重物掉落在铁砧上,物体的质量通过弹簧由铁砧传递给放置于合成材料面
层测试样品上的一个弧形底座测力台。测力台装有一个压力传感器,它能记录撞击时的最高压力。将
这个力的最高值与在混凝土表面上测得的结果进行对比,计算合成材料表面冲击吸收百分比。
测试在混凝土表面的读数
C
F 时,将检测仪器垂直地放置在混凝土地板上,将下落重物的下端调整
到距离铁砧(55±0.25)mm 的正上方,释放下落重物,使其自由落体砸在铁砧上。记录在冲击过程中,
混凝土表面所受到的冲击力的峰值数值。再重复上述的测试过程 10 次,使总测试次数达到 11 次。记
录下第二次到第十一次冲击力峰值的平均值,作为
C
F ,并记录在软件中。
C
F 的数值应该在(6.60±
0.25)kN 的范围内,如果数值超出这个数据,测试结果将被视为无效。通常
C
F 的测试每三个月要做一
次,作为仪器及测试环境的校准测试。
在实验室测试合成材料样品冲击吸收值时,应该对样品在 3 个温度点 0 ℃±2 ℃,23 ℃±2 ℃
,50 ℃±2 ℃下进行测试。
测试在合成材料表面的读数
S
F 时,将检测仪器垂直地放置在测试样品上,将下落重物的下端调
整到距离铁砧(55±0.25)mm 的正上方,释放下落重物,使其自由落体砸在铁砧上。记录在冲击过程
中,合成材料表面所受到的冲击力的峰值数值。经过第一次测试后,间隔(60±10)s 进行第二次测试。
经过冲击表面后,为了不让合成材料表面负重太久,应在几秒钟内从铁砧上提起重物。
从 0 ℃温度点开始依序往高温度点测试样品时,先将待测样品放置在-5 ℃冰箱中恒温 1 h,然
后将样品取出放置到测试平台待测,当样品表面温度自然上升至 0 ℃±2 ℃、23 ℃±2 ℃时,分别
开始测试,每一个温度点的测试要在 5 min内完成,每一个点位测试 3 次,取后 2 次数值计算冲击吸
收值,结果取其算术平均值,为该点位在相应温度点的测定值。
从 50 ℃温度点开始依序往低温度点测试样品时,先将待测样品放置在 55 ℃烘箱中恒温 1h,然
后将样品取出放置到测试平台待测,当样品表面温度自然下降至 50 ℃±2 ℃时,开始测试,每一个
温度点测试要在 5 min内完成,每一个点位测试 3 次,取后 2 次数值计算冲击吸收值,结果取其算术
平均值,为该点位在相应温度点的测定值。
样品的表面温度,使用经校准后的红外线测温仪测量。
D.3 结果计算
冲击吸收按式(D.1)进行计算,数值精确到小数点后一位。

附 录 E
(规范性附录)
合成材料面层垂直变形的检测方法
E.1 检测装置
E.1.1 采用垂直变形测试仪(见图 E.1 所示)检测合成材料跑道面层的垂直变形性能。
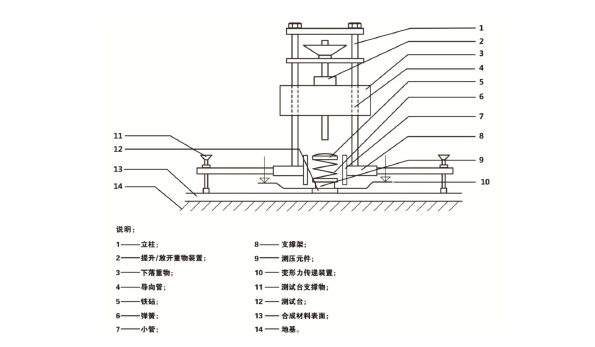
图 E.1 垂直变形测试仪
图 E.1 垂直变形测试仪
E.1.2 垂直变形测试仪及地基应符合下列要求:
a) 下落重物的质量为(20±0.1)kg,并有一个坚硬光滑的表面,使其可以以最小的摩擦力垂直通
过导向管无阻碍的落下;
b) 螺旋弹簧直径为(69±1)mm,上层为硬化表面,在0.1 kN到1.6 kN的范围内,有着(40±1.5)N/mm
的线性弹簧弹性度;
c) 可调节测力台支撑物,距离点弹性运动面层的测试点最少 250mm;距离面弹性运动面层的测试
点最少 600mm;
d) 钢制测力台,其底盘下面层呈平面状,边缘半径为 1mm,底盘直径为(70.0±0.1)mm,厚度最
少为 10mm;
e) 金属导向管,其内沿内径是(71.0±0.1) mm;
f) 测试脚,是由钢制测力台,压力传感器,弹簧和铁砧(最低厚度 20mm,以盘中心的测量值为
准)组成的,固定在金属导向管中,整体质量(不计导向管)应该为(3.5±0.35)kg;
g) 提升与释放重物装置,可以让其从设定的高度跌落,且误差不大于±0.25 mm;
h) 测试形变用的传感器可采用测试范围为±10 mm,误差不超过 0.05 mm 的电子变形传感器。传
感器距离整个测试仪器的中轴线的距离应该≤125 mm。两个感应器应该以仪器的中轴线对称放
置在变形力传递装置上;
i) 具有记录和调节放大从压力传感器输出的信号的装置以及显示这些记录的装置。放大器的频道
频率应该大于等于 1 kHz;
j) 测试地基应该是一块平整、坚硬、无振动的混凝土地板。
E.2 检测方法
测试在合成材料样品垂直变形值时,将检测仪器垂直地放置在合成材料样品上,将下落重物的下
端调整到距离铁砧(120±0.25) mm的正上方,释放下落重物,使其自由落体砸在铁砧上。记录在冲击
过程中,合成材料表面所发生的形变数值。经过一次测试后,间隔(60±10) s进行二次测试。经过冲
击表面后,为了不让合成材料表面负重太久,应在几秒钟内从铁砧上提起重物。每一个点位测试 3 次,
取后 2 次数值计算垂直变形值,结果取其算术平均值,数值精确到小数点后一位。
E.3 结果表示
垂直变形是根据 1500 N 动力冲击测试中,超过 400 N 的读数结果计算得出的。测试结果是最后两
次冲击的平均值,数值精确到小数点后一位。
附 录 F
(规范性附录)
合成材料面层抗滑值的检测方法
F.1 检测装置
采用图 F.1 中所示滑动阻力测试仪进行检测。
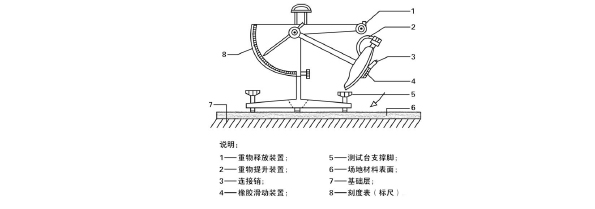
图 F.1 便携式阻力测试仪
F.2 检测方法
将一个标准的光滑橡胶滑动片安装在摆动臂末端的支撑块之下,并用弹簧顶住。这个滑动片将随摆
动臂从 90°位置向下摆向样品表面,并沿着表面滑动一定距离,摆动臂摆动时带动一个惰性指针,使
指针停留在摆动的最高点位置上。
将滑动阻力测试仪水平放置在样品表面,放开撑脚,以防止当摆动臂摆过表面时,支撑脚下方合成
材料的表面出现局部偏斜。当摆动臂从正常的水平位置自由下落时,指针停留的刻度应是零点,否则,
应调节摩擦环(在摆动臂的定位中心处)并反复操作,直到始终得到一个零点。
测试样品时,调节摆动臂的高度,使滑动片与被测表面接触,滑动片从左边缘到右边缘与被测表面
接触的距离是在 125 mm~127 mm 之间。把所设置的高度固定在这个位置上并反复摆动滑动片以核定距
离。然后,把摆动臂放在水平重物的位置上。
在测试区保持测试样品表面干燥,放开摆动臂使其自由落下,略去第一次指针计数,然后进行 5
次同样的试验。记录每次摆动后指针所得的刻度读数,计算这 5 个读数的平均值,数值精确到小数点后
一位,即为干燥表面的抗滑值。
在测试区洒上干净的水,放开摆动臂使其自由落下,略去第一次指针计数,然后进行 5 次同样的试
验。记录每次摆动后指针所得的刻度读数,计算这 5 个读数的平均值,数值精确到小数点后一位,即为
潮湿表面的抗滑值。
如果合成材料表面显示具有方向性的图案,那么,用仪器应能测出各个方向不同的数值。方法是调
节仪器,使滑动部件从开始摆动方向的 90°和 180°通过相同的一块表面,所测得结果可作为第一组读
数的参考数。
附 录 G
(规范性附录)
短链氯化石蜡(C 10 -C 13 )含量的测试 气相色谱-电子捕获负化学电离源质谱法
G.1 概述
本方法规定了用气相色谱-质谱法测定合成材料面层成品及其原料中短链氯化石蜡(C 10 -C 13 )的含
量的方法。
G.2 原理
样品采用正己烷或其他合适的溶剂作为萃取溶剂进行超声萃取,所得试样溶液经浓硫酸净化处理
后,用气相色谱-电子捕获化学电离源质谱仪进行定性定量分析。
本方法详细给出了内标法定量的测试步骤,实验室也可以经方法确认后采用外标法进行定量分析。
G.3 试剂和材料
G.3.1 萃取溶剂
正己烷或其他合适的溶剂,分析纯。
G.3.2 内标标准物质
可选用ε -六六六(CAS:6108-10-7,100 mg/L)、1,1,1,3,10,11-六氯十一烷(CAS:601523-28-8,100
mg/L),或其他合适的内标物质。
G.3.3 短链氯化石蜡(C 10 -C 13 )标准储备溶液
51.5%平均氯化度,100 mg/L。
55.5%平均氯化度,100 mg/L。
63.0%平均氯化度,100 mg/L。
G.3.4 浓硫酸
分析纯。
G.4 仪器和设备
G.4.1 气相色谱-质谱联用仪:配有电子捕获负化学电离源。
G.4.2 超声波发生器。
G.4.3 电子天平:精确度 0.1 mg。
G.4.4 离心机:转速 5000 r/min~20000 r/min。
G.5 分析步骤
G.5.1 短链氯化石蜡(C 10 -C 13 )标准工作溶液
将平均氯化度为 51.5%、55.5%和 63.0%的短链氯化石蜡(C 10 -C 13 )标准溶液等比例混合,充分混匀
后,用萃取溶剂(G.3.1)将其配制成 5 mg/L、10 mg/L、15 mg/L、20 mg/L、25 mg/L、50 mg/L 的系
列标准工作溶液,其中内标浓度为 5 mg/L。根据需要可配制成其他合适的浓度。
G.5.2 超声波萃取
称取约 0.5 g 样品,精确至 0.1 mg,放入于螺口刻度试管(带密封盖),加入 1 mL 内标(G.3.2)
和 9 mL 萃取溶剂(G.3.1),用超声波发生器在 60℃水温下超声萃取 60 min,摇匀冷却后待处理。
注1:高浓度样品可进行两次萃取测试。
注2:如果试样溶液中有颗粒物质,取一部分试液经0.45 μ m有机系微孔滤膜过滤后用于检测分析。
G.5.3 净化
取 5 mL 试样溶液于玻璃管,并加入 5 mL 98%浓硫酸,混匀并用离心机分离,收集上层有机溶液。
重复以上操作直至下层酸溶液澄清或至白色,此过程不超过五次。
此溶液可依据其实际情况直接进样,或者用萃取溶剂(G.3.1)稀释后测试。
G.5.4 气相色谱-质谱分析条件
由于测试结果取决于所使用的仪器,因此不可能给出仪器分析的普适参数,可参考的仪器条件
如下:
a) 色谱柱:DB-5MS 毛细管柱,30 m×0.25 mm×0.1 µm,或其他具有相当性质的毛细管柱;
b) 进样口温度:300 ℃;
c) 柱温:程序升温,80 ℃保持以 40 ℃/min 升至 300 ℃保持 5 min;
d) 质谱接口温度:280 ℃;
e) 离子源温度:200 ℃;
f) 质量扫描范围:50 amu~550amu;
g) 进样方式:不分流进样;
h) 电离方式:电子捕获负化学源电离(ECNI);
i) 载气:氦气(99.999%),流量为 2.0 mL/min;
j) 反应气:甲烷,流速为 1.5 mL/min;
k) 进样量:1.0 µL;
l) 溶剂延迟:2.0 min。
G.5.5 试验
分别取 1 µL 标准工作溶液与试样溶液注入色谱仪,按分析条件操作。通过比较试样与标样的保留
时间及特征离子进行定性或定量分析。
G.5.6 定性定量分析
根据各实验室仪器所适合的分析条件对标准溶液及试样溶液进行分析,根据色谱峰的保留时间和特
征离子的相对丰度进行定性分析,在确认是短链氯化石蜡(C 10 -C 13 )的条件下,采用定量离子进行定量
分析。
定量分析参考表 G.1中的定量离子,采用内标法定量。
表 G.1 短链氯化石蜡(C 10 -C 13 )的定性参考离子和定量选择离子
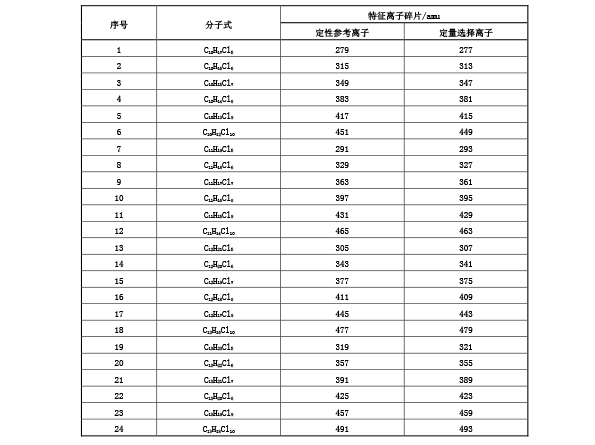
G.5.7 空白试验
除不加试样外,均按上述分析步骤进行。

G.7 检出限
本方法短链氯化石蜡(C 10 -C 13 )含量的参考检出限为0.1 g/kg。
G.8 精密度
同一实验室相同条件下,在短时间内对同一被测对象,两次独立测定结果的绝对差值不应大于其算
术平均值的 15%。
附 录 H
(规范性附录)
4,4'-二氨基-3,3'-二氯二苯甲烷(MOCA)含量的测试
H.1 概述
本方法规定了用气相色谱-质谱法和高效液相色谱法测定合成材料面层中 MOCA 含量的方法,其中气
相色谱-质谱法为仲裁法。
H.2 方法一:气相色谱-质谱法(仲裁法)
H.2.1 原理
用适当的溶剂萃取试样,萃取液经过滤后加入蒽-d 10 作为内标物,取适量样液用气相色谱-质谱联
用仪进行分析,用内标法计算试样溶液中 MOCA 的含量。
本方法详细给出了内标法定量的测试步骤,实验室也可以经方法确认后采用外标法进行定量分析。
H.2.2 材料和试剂
H.2.2.1 萃取溶剂
丙酮或其他合适的溶剂,分析纯。
H.2.2.2 内标溶液
用萃取溶剂(H.2.2.1)将蒽-d 10 (CAS No.:1719-06-8)配制成浓度约为 50mg/L 的内标溶液。
H.2.2.3 MOCA (CAS No.:101-14-4)标准样品
纯度≥99%或已知纯度。
H.2.2.4 标准工作溶液
用萃取溶剂(H.2.2.1)将 MOCA(H.2.2.3)配制成 5 mg/L、10 mg/L、25 mg/L、50 mg/L 的标准
工作溶液。
注:标准工作溶液现配现用,根据需要可配制成其它合适的浓度。
H.2.2.5 混合标准溶液
移取 1 mL 标准溶液(H.2.2.4),加入 100 μ L 内标物溶液(H.2.2.2),配制成混合标准溶液。
H.2.3 仪器设备
H.2.3.1 气相色谱-质谱联用仪。
H.2.3.2 超声波发生器。
H.2.3.3 电子天平:精确度 0.1 mg。
H.2.4 分析步骤
H.2.4.1 样品制备
称取 0.5g 样品,精确至 0.1mg,放入螺口刻度试管(带密封盖),加入 10mL 萃取溶剂(H.2.2.1)
并密封试管,置于超声波发生器(H.2.3.2)中,超声萃取 60min。萃取完成后,取出刻度试管冷却至
室温并混合均匀。此试样溶液可依据其实际情况直接进样,或者用萃取溶剂(H.2.2.1)稀释后进行分
析。
移取 1 mL 上述待测液至色谱配样瓶中,加入 100 μ L 内标溶液(H.2.2.2)混匀后进行气相色谱-
质谱分析。
注 1:高浓度样品可进行两次萃取测试。
注 2:如果试样溶液中有颗粒物质,取一部分试液经 0.45μ m 有机系微孔滤膜过滤后用于检测分析。
H.2.4.2 内标标准曲线的绘制
对标准混合溶液(H.2.2.5)进行气相色谱-质谱分析,以待测物的质量浓度为横坐标、MOCA 和对
应内标物峰面积的比值为纵坐标作图,可得一条通过原点的直线,即内标标准曲线。
H.2.4.3 气相色谱-质谱分析条件
由于测试结果取决于所使用的仪器,因此不可能给出仪器分析的普适参数,可参考的仪器条件如下:
a) 色谱柱(基本柱):聚二甲基硅氧烷毛细管柱,30 m×0.32 mm×1.0 μ m;
b) 进样口温度:250 ℃;
c) 柱温:程序升温,35 ℃保持 4m in,然后以 8 ℃/min 升至 300 ℃保持 10 min;
d) 质谱接口温度:270 ℃;
e) 离子源温度:230 ℃;
f) 质量扫描范围:35 amu~350 amu;
g) 进样方式:不分流进样;
h) 载气:氦气(纯度≥99.999%),流量为 1.0 mL/min;
i) 进样量:1.0 μ L;
j) 离子化方式:EI;
k) 离子化电压:70 eV;
l) 溶剂延迟:3.0 min。
H.2.4.4 定性定量分析
分别取 1 μ L 标准工作溶液(H.2.2.4)与试样溶液按 H.2.4.3 条件进行气相色谱-质谱分析。通过
比较试样与标样的保留时间及特征离子进行定性。必要时,选用另外一种或多种方法对异构体进行确认。
移取 1 mL 稀释后的试样溶液,加入 100 μ L 内标溶液(H.2.2.2)混匀后待测。然后分别取 1 μ L
混合标准溶液(H.2.2.5)与待测溶液按 H.2.4.3 条件进行气相色谱-质谱分析,选用选择离子方式进行
定量。
定量分析可参考表 H.1 中的定量离子。
表 H.1 MOCA 与内标物的定性参考离子和定量选择离子

m ——试样的质量,单位为克(g)。
注:待测液中 MOCA 的含量的也可用外标法计算。
H.2.6 检出限
本方法 MOCA 的参考检出含量为0.01 g/kg。
H.2.7 精密度
同一实验室相同条件下,在短时间内对同一被测对象,两次独立测定结果的绝对差值不应大于其算
术平均值的 15%。
H.3 方法二:高效液相色谱法
H.3.1 原理
用适当溶剂萃取试样,经超声水浴萃取后静置冷却至室温,取适量试样溶液过微孔滤膜,用高效液
相色谱仪进行分析,外标法计算试样溶液中 MOCA 的含量。
H.3.2 材料和试剂
H.3.2.1 甲醇
色谱纯。
H.3.2.2 水
GB/T 6682-2008推荐使用的二级水。
H.3.2.3 萃取溶剂
丙酮或其他合适的溶剂,色谱纯。
H.3.2.4 MOCA (CAS No.:101-14-4)标准样品
纯度≥99%或已知纯度。
H.3.2.5 MOCA 储备液
准确称取 MOCA 标准物质(H.3.2.4)50 mg(精确到 0.1 mg)于 100 mL 容量瓶中,用甲醇(H.3.2.1)
溶解并稀释到刻度,摇匀,即得到浓度为 500 μ g/mL MOCA 标准储备液。
H.3.2.6 MOCA 系列标准工作溶液
准确量取适当体积的 MOCA 储备液(H.3.2.5)于 10 mL 容量瓶中,用萃取溶剂(H.3.2.3)稀释并
定容至刻度,得到浓度为 1 μ g/mL、2 μ g/mL、5 μ g/mL、10 μ g/mL、20 μ g/mL 和 50 μ g/mL MOCA
系列标准溶液。
H.3.2.7 有机相微孔滤膜
孔径0.22 μ m。
H.3.3 仪器与设备
H.3.3.1 高效液相色谱仪,配有 UV-Vis 检测器。
H.3.3.2 超声波发生器。
H.3.3.3 电子天平,精确度 0.1 mg。
H.3.3.4 高速离心机,转速不小于 10000 r/min。
H.3.4 分析步骤
H.3.4.1 样品制备
称取约0.5 g 样品,精确至0.1 mg,置于50 mL 带密封盖的玻璃瓶中,准确加入10.0 mL 甲醇+丙酮
(v+v=1+1)混合溶液,超声萃取60 min,得到试样溶液,将试样溶液静置冷却至室温,如有必要可将
试样溶液以10000 r/min 的转速离心10 min,取离心后的上层清液过0.22 µm 微孔滤膜(H.3.2.7)后,
滤液作为待测液。
此试样溶液可依据其实际情况直接进样,或者用萃取溶剂(H.3.2.3)稀释后进行分析。
H.3.4.2 高效液相色谱条件
由于测试结果取决于所使用的仪器,因此不可能给出仪器分析的普适参数,可参考的仪器条件如下:
a) 色谱柱:C18反相柱(250 mm×4.6 mm(i.d.)×5 μ m)或相当者;
b) 流动相:甲醇+水溶液=70+30(体积比);
c) 流速:1.0 mL/min;
d) 柱温:30 ℃;
e) 进样体积:20 μ L;
f) 检测波长:254 nm。
H.3.4.3 定性定量分析
参照测试条件,待仪器稳定后,对制备的样品溶液进行测定,本方法采用色谱峰的保留时间和光
谱图进行定性分析。如果检出的色谱峰的保留时间与光谱图和标准品一致,则可判断样品中有 MOCA
的存在。
采用外标法进行定量分析,根据 H.3.2.6配制的 MOCA 系列标准工作溶液,将该系列标准工作溶
液和待测液在相同条件下进样分析。以标准溶液的浓度为横坐标、MOCA 峰面积为纵坐标做图,得到
标准曲线回归方程。将样品中检测出的 MOCA 的峰面积代入标准曲线,即可得样品溶液中 MOCA 的质
量浓度,将其代入式(H.3)即可计算出样品中 MOCA 的含量。
H.3.4.4 空白试验
除不加试样外,均按上述分析步骤进行。

H.3.6 检出限
本方法 MOCA 的参考检出限为 0.001 g/kg。
H.3.7 精密度
同一实验室相同条件下,在短时间内对同一被测对象,两次独立测定结果的绝对差值不应大于其算
术平均值的 15%。
附 录 I
(规范性附录)
有害物质释放量的测试 小型环境测试舱法
I.1 概述
本方法规定了用小型环境测试舱测定合成材料面层有害物质释放量的方法。
I.2 原理
将试样置于指定温度、湿度和通风条件的环境测试舱中,经过一定的平衡时间之后通过检测舱内空
气中有害物质浓度确定试样的有害物质释放量。
I.3 小型环境测试舱
小型环境测试舱由有效容积为 60 L 的密封舱、空气净化系统、空气温湿度调节控制系统、空气温
湿度监控系统、空气流量调节控制装置、空气采样系统等部分组成。如图 I.1 所示。
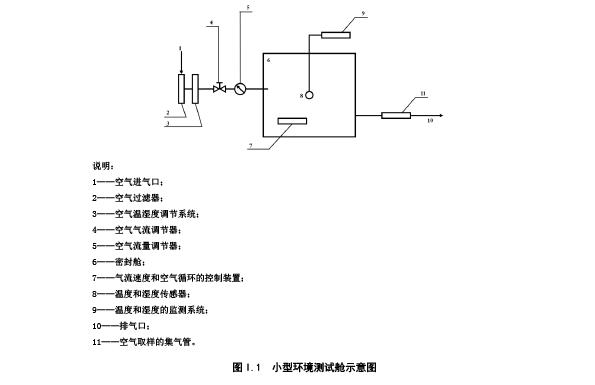
I.4 环境测试舱舱内试验条件
环境测试舱舱内试验条件如下:
——空气温度(60±2) ℃;
——空气相对湿度(5±2) %;
——空气交换速率(1±0.01)h
-1 ;
——试样表面空气流速 0.1 m/s~0.3 m/s;
——材料/舱载荷比 0.4 m
2 /m 3 。
I.5 试样的制备
I.5.1 受检试样到达实验室后试样应存放在温度(25±5)℃的室内环境带包装保存,并应尽快检验。
I.5.2 试样制备时,试验环境保持清洁通风,避免试样被污染。
I.5.3 合成材料面层试样应从距样品边缘至少 20 mm 处按要求的面积截取,并将试样的人为切割表面
及底面用铝箔包覆。
I.5.4 以试样的上表面曝露面积进行计算,保证其材料/舱负荷比为 0.4 m
2 /m 3 。
I.5.5 试样制备后应将其置于温度(23±2)℃,相对湿度(50±10)%的无污染环境中进行 24h 的预
平衡,之后置于环境测试舱内进行测试。
I.6 测试步骤
I.6.1 环境测试舱的准备
I.6.1.1 试验前对环境测试舱进行清洗。首先用碱性清洗剂(pH 值≥7.5)清洗舱内壁,再用去离子
水或蒸馏水擦洗舱内壁,敞开舱门,开启风扇至舱体风干。
I.6.1.2 在环境测试舱运行六次换气之后,通过空气采样检测环境测试舱的背景浓度。
I.6.1.3 当舱内甲醛背景浓度不大于 6 μ g/m
3 ,总挥发性有机化合物(TVOC)背景浓度不大于 50 μ
g/m
3 ,其他单一污染物本底浓度不大于 5 μ g/m 3 时方可进行进一步测试。
I.6.2 试样的平衡
I.6.2.1 将按 I.5 制备的试样放入环境测试舱居中位置,散发面应水平向上,使空气气流均匀地从试
件表面通过,并迅速关闭环境测试舱舱门开始试验。
I.6.2.2 以试件放入环境测试舱的时刻为 0 时刻计。
I.6.3 舱内空气采样分析
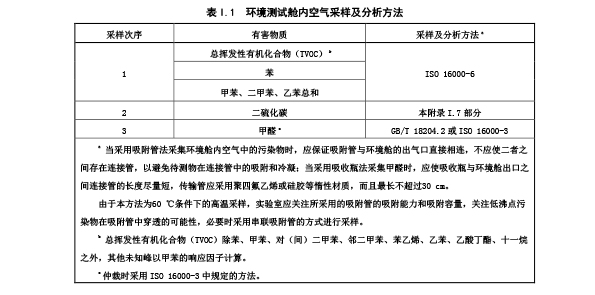
I.6.3.1 试样在环境测试舱内平衡(24±1) h 之后进行舱内空气采样分析。
I.6.3.2 按照表 I.1 规定的先后次序进行舱内空气采样,采样流速不应大于环境舱供给气流的 80%。
I.6.3.3 舱内空气采样及分析方法见表 I.1。
I.7 空气中二硫化碳的热解吸测试方法
I.7.1 原理
用活性炭管采集环境舱空气中的 CS 2 ,然后用热解吸仪解吸,用气相色谱仪或气相色谱质谱联用仪
分析,保留时间定性,峰面积定量。
I.7.2 仪器设备
I.7.2.1 活性炭采样管
用与热解吸匹配的不锈钢管,装入 200 mg 活性炭,两端用少量玻璃棉固定,装好后的管用纯氮气
于(300~350) ℃温度条件下吹扫 1 h,然后套上塑料帽封紧管的两端,干燥器中可保存 5 d。
I.7.2.2 恒流大气采样器
流量范围(0.02~0.5) L/min,流量稳定,使用时用流量计校准采样系统在采样前和采样后的流
量。流量误差小于 5%。
I.7.2.3 气相色谱仪
配备火焰光度检测器(394 nm 硫滤光片)、质谱检测器或其他合适检测器。
I.7.3 采样和样品保存
在采样地点打开活性炭采样管,使其与环境舱出气口直接相连,以 0.2 L/min~0.5 L/min 的流速,
抽取(1~10)L 舱内空气。采样后,将管的两端套上塑料帽,记录采样时的温度和大气压力,尽快拿
到实验室分析。
I.7.4 分析步骤
I.7.4.1 样品解吸条件
可参考的解吸条件如下:
a) 解吸温度:300 ℃;
b) 解吸时间:10 min;
c) 解吸气流量:(30~50)mL/min;
d) 冷阱制冷温度:2 ℃;
e) 冷阱加热温度:280 ℃。
I.7.4.2 色谱分析条件
可参考的色谱分析条件如下:
a) 色谱柱:DB-5,60 m×0.25 mm×0.25 μ m 毛细管柱或其他等效色谱柱;
b) 升温程序:初始温度 50 ℃保持 5 min,以 25 ℃/min 的速率升至 250 ℃,保持 10 min;
c) 进样口温度:250 ℃;
d) 其它仪器条件根据使用的检测器的不同自行确认。
I.7.4.3 标准曲线的绘制
标准曲线的绘制分为气体外标法和液体外标法。
a)气体外标法
用泵准确抽取浓度约 10 mg/m
3 的标准气体,100 mL、200 mL、400 mL、1 L、2 L 通过吸附管,为
标准系列。
b)液体外标法
用液体外标法制备注射装置分别取(0.5~10)μ L,含液体组分约 2000 mg/L 的标准溶液注入活性
炭吸附管,同时用 100 mL/min 的惰性气体通过活性炭吸附管,5 min 后取下吸附管密封,为标准系列。
二硫化碳标准溶液配制的参考步骤:在25 mL容量瓶中加入无水乙醇(或甲醇)约15 mL,盖塞
称重(精确至0.1 mg),然后加入二硫化碳(色谱纯)约50 mg(精确至0.1 mg)。用无水乙醇(或
甲醇)稀释至标线,计算其中二硫化碳的准确浓度。
I.7.4.4 样品分析
每支样品吸附管按绘制标准曲线的操作步骤进行分析。
I.8 结果的计算
I.8.1 舱内有害物质浓度的标准化
所采空气样品中各组分有害物质浓度按式(I.1)换算成标准状态下的浓度:

I.9 检出限
按上述方法测试总挥发性有机化合物(TVOC),苯,甲苯、二甲苯和乙苯总和,二硫化碳,甲醛的
释放量,其检出限不应大于该项指标的 1/10,由实验室确定。
附 录 J
(规范性附录)
气味评定
J.1 试验装置和评定小组
J.1.1 测试瓶:测试瓶的容量为 1 L。带有可密闭的盖子,在室温或 60 ℃下应是无气味的。
J.1.2 恒温箱:空气循环型,温度波动不大于±2 ℃。
J.1.3 气味评定实验室应符合 HJ 865-2017 的建设要求,并要求环境无气味。
J.1.4 气味评定小组:为了获得一致的数据,气味评定小组应由不少于 5 名,年龄在 18 岁~45 岁,
不吸烟、嗅觉器官无疾病,并经嗅觉检测,具有气味评定资质的气味评定人员组成。评定当天,气味评
定人员不应携带和使用带气味的香料、化妆品或护肤品,不应食用有刺激气味的食物。患感冒或嗅觉器
官不适的气味评定人员不应参加当天的测定。
J.2 样品制备
对于合成材料面层成品,从距样品边缘至少 20 mm 处截取规格为 20 mm×50 mm×实际厚度的试样,
用铝箔包覆试样的侧面及底面;对于人造草面层填充颗粒,直接取样 20 g。
J.3 气味评定
将取好的样品放入 1 L 的测试瓶内,测试瓶在 60 ℃恒温箱中保持 2h,待冷却到室温进行气味评定,
测试瓶从恒温箱中取出到评定应在 0.5 h 内完成。
评定时,气味评定人员应把鼻子靠近测试瓶口,然后移去盖子,立即吸入瓶内气体。如果需要重复
测试,应在容器被再次打开前关闭 2 min。每个测试瓶内的气体样品最多可供 3 名气味评定人员进行测
试;每个气味评定人员只能对一组气味评定实验进行一次气味评定。
为了避免适应性效应,气味评定人员应在 2 次测定间暂停不少于 2 min。为避免嗅觉疲劳,1 h 内
连续测定次数不应超过 5 次。
J.4 气味评定等级
气味评定人员评定及记录样品气味等级。气味等级见表 J.1。
表 J.1 气味等级

J.5 结果表述
气味评定等级尽量用整数表示,必要时也可使用半数表示。
某个评定结果与所有评定结果中位数相差 1.5 或更多,则为无效评定结果;如果存在两个或两个以
上无效评定结果,或者有效评定结果少于 5 个,则应重新进行评定。
取所有有效评定结果的中位数作为气味评定等级值,结果保留至小数点后一位。
附 录 K
(规范性附录)
合成材料场地现场取样位置的确定规则
K.1 原理
以运动场地为一个平面,以其相邻两边为坐标轴,通过产生随机数的方式确定取样坐标。
K.2 器材
K.2.1 卷尺:精确到 1 cm。
K.2.2 随机数骰子:符合 GB/T 10111 的规定。
K.2.3 绳子等辅助工具。
K.3 试验方法
K.3.1 测量运动场地的长度 L、宽度 W,选取运动场地的任意相邻两边中长边为横坐标轴,短边为纵坐
标轴,规定横坐标代表运动场地中某点到纵坐标轴的垂线段的长度。
K.3.2 不规则的运动场地则先划定包含场地的最小矩形,然后进行长度和宽度的测量及坐标轴的确定。
K.3.3 按照 GB/T 10111-2008 中 5.2.2 的规定,用两个随机数筛子操作一次,产生一个 1~99 的随机
数 R,若得的随机数是 00 则重新操作。
K.3.4 按照公式(K.1)计算取样位置中心点的横坐标。
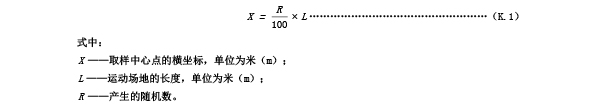
K.3.5 根据公式(K.1)产生的横坐标确定一条平行于纵坐标的线段,取样区域为该线段与合成材料跑
道重叠部分中适宜取样的区域。
K.3.6 挖取样品中心点应落于 K.3.5 产生的线段上,样品边缘距合成材料场地边缘距离至少 10 cm,
若挖取样品不是正方形,应保证矩形样品的长边与运动场地的长边平行。
K.3.7 若上述随机抽取的位置出现未浇筑合成材料面层或距离面层边缘过近等不适于取样的情况,则
重复上述 K.3.4~K.3.6 的过程,直到确定合适的取样位置。
K.3.8 如有必要,用上述规则抽取第 2 块或更多样品,但两个取样中心点之间的直线距离不应小于 10
m,否则应重复上述 K.3.4~K.3.6 的过程,直到确定合适的取样位置。
参 考 文 献
[1] GB 18582-2008 室内装饰装修材料 内墙涂料中有害物质限量
[2] GB 19272-2011 室外健身器材的安全 通用要求
[3] GB/T 20033.3-2006 人工材料体育场地使用要求及检验方法 第 3 部分:足球场地人造草面层
[4] GB/T 22753-2008 玩具表面涂层技术条件
[5] GB/T 29614-2013 硫化橡胶中多环芳烃含量的测定
[6] GB 30982-2014 建筑胶粘剂有害物质限量
[7] GB 50325-2010 民用建筑工程室内环境污染控制规范
[8] HG/T 4065-2008 胶粘剂气味评价方法
[9] SJG 29-2016 合成材料运动场地面层质量控制标准
[10] SN/T 2570-2010 皮革中短链氯化石蜡残留检测方法 气相色谱法
[11] T/310101002-C003-2016 学校运动场地塑胶面层有害物质限量
[12] 中国田径协会.国际田径协会联合会田径场地设施标准手册(2008 年版)[M].北京:人民体育
出版社,2009(2017.4.重印).
[13] ISO 16000-9:2006 Indoor air - Part 9: Determination of the emission of volatile
organic compounds from building products and furnishing -- Emission test chamber
method
[14] DIN 18035-6:2014 运动场地-第 6 部分:合成材料地面
[15] EN 71-3:2013 Safety of toys Part 3: Migration of certain elements
[16] EN 14877:2013 Synthetic surfaces for outdoor sports areas – Specification
返回顶部▲
24小时咨询热线:吴总18520207988